Reverse objects
reverse.RdReverses objects, mainly meant for dna_seg and comparison objects.
Usage
reverse(x, ...)
# Default S3 method
reverse(x, ...)
# S3 method for class 'dna_seg'
reverse(x, ...)
# S3 method for class 'comparison'
reverse(x, side = 0, ...)Examples
## load data
data(three_genes)
dna_segs <- three_genes$dna_segs
comparisons <- three_genes$comparisons
## on dna_seg
dna_segs[[1]]
#> name start end strand fill gene_type region_plot col lty lwd
#> <char> <num> <num> <num> <char> <char> <char> <char> <num> <num>
#> 1: feat1 2 600 -1 blue arrows NA gray20 1 1
#> 2: feat2 1000 800 -1 grey arrows NA gray20 1 1
#> 3: feat3 1050 1345 1 red arrows NA gray20 1 1
#> pch cex
#> <num> <num>
#> 1: 8 1
#> 2: 8 1
#> 3: 8 1
reverse(dna_segs[[1]])
#> name start end strand fill gene_type region_plot col lty lwd
#> <char> <num> <num> <num> <char> <char> <char> <char> <num> <num>
#> 1: feat1 -600 -2 1 blue arrows NA gray20 1 1
#> 2: feat2 -800 -1000 1 grey arrows NA gray20 1 1
#> 3: feat3 -1345 -1050 -1 red arrows NA gray20 1 1
#> pch cex
#> <num> <num>
#> 1: 8 1
#> 2: 8 1
#> 3: 8 1
## on comparison
reverse(comparisons[[2]], side = 1)
#> start1 end1 start2 end2 col direction
#> <num> <num> <num> <num> <char> <num>
#> 1: -50 -500 1899 2034 #67000D -1
#> 2: -800 -1100 2732 2508 #08306B 1
reverse(comparisons[[2]], side = 3)
#> start1 end1 start2 end2 col direction
#> <num> <num> <num> <num> <char> <num>
#> 1: -50 -500 -1899 -2034 #67000D 1
#> 2: -800 -1100 -2732 -2508 #08306B -1
## With mauve backbone
data(mauve_bbone)
## Plot
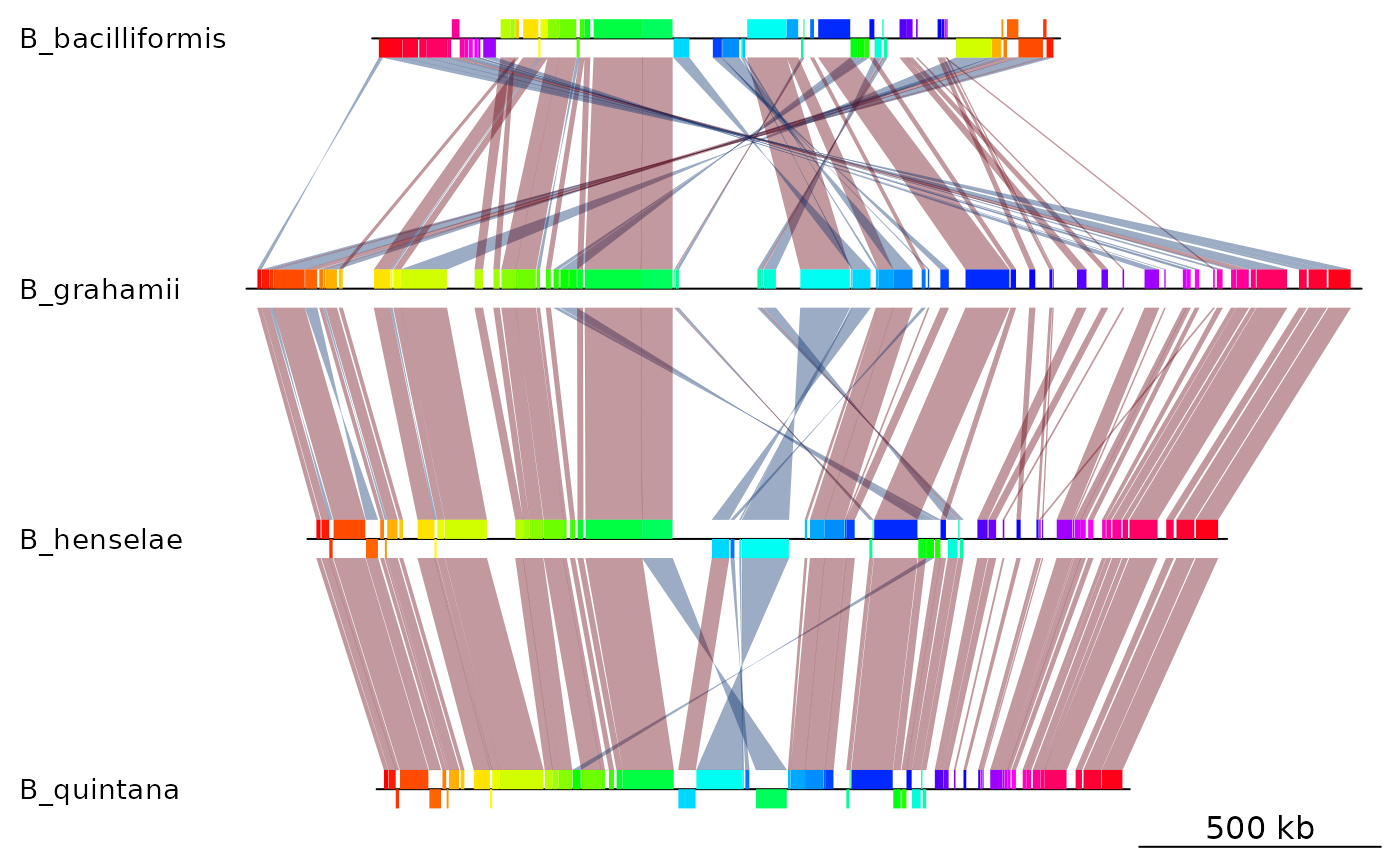
plot_gene_map(dna_segs = mauve_bbone$dna_segs,
comparisons = mauve_bbone$comparisons,
alpha_comparisons = 0.4)
 ## Reverse B_bacilliformis, and the corresponding comparison (first "side")
mauve_bbone$dna_segs[[1]] <- reverse(mauve_bbone$dna_segs[[1]])
mauve_bbone$comparisons[[1]] <- reverse(mauve_bbone$comparisons[[1]], 1)
plot_gene_map(dna_segs = mauve_bbone$dna_segs,
comparisons = mauve_bbone$comparisons,
alpha_comparisons = 0.4)
## Reverse B_bacilliformis, and the corresponding comparison (first "side")
mauve_bbone$dna_segs[[1]] <- reverse(mauve_bbone$dna_segs[[1]])
mauve_bbone$comparisons[[1]] <- reverse(mauve_bbone$comparisons[[1]], 1)
plot_gene_map(dna_segs = mauve_bbone$dna_segs,
comparisons = mauve_bbone$comparisons,
alpha_comparisons = 0.4)
